This page lists questions that marketing-authorisation holders (MAHs) may have on type-IA variations. It provides an overview of the European Medicines Agency's position on issues that are typically addressed in discussions or meetings with MAHs in the post-authorisation phase. Revised topics are marked 'New' or 'Rev.' upon publication.
A PDF version of the entire post-authorisation guidance is available:
European Medicines Agency post-authorisation procedural advice for users of the centralised procedure
Reference Number: EMEA-H-19984/03 Rev. 108English (EN) (2.65 MB - PDF)
First published: 01/07/2009 Last updated: 21/06/2024European Medicines Agency post-authorisation procedural advice for users of the centralised procedure: document with track changes
Reference Number: EMEA-H-19984/03 Rev. 108English (EN) (2.65 MB - PDF)
First published: 09/12/2013 Last updated: 21/06/2024These questions and answers have been produced for guidance only and should be read in conjunction with the rules governing medicinal products in the European Union, volume 2, notice to applicants.
MAHs must in all cases comply with the requirements of Community legislation. Provisions that extend to Iceland, Liechtenstein and Norway by virtue of the European Economic Area agreement are outlined in the relevant sections of the text.
Commission Regulation (EC) No 1234/2008 ('the Variations Regulation') and the Commission guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 and on the documentation to be submitted pursuant to those procedures ('the Variations Guidelines') set out a list of changes to be considered as type-IA variations. Such minor variations have only a minimal impact or no impact at all, on the quality, safety or efficacy of the medicinal product, and do not require prior approval before implementation ('do-and-tell' procedure). The Classification Guideline clarifies the conditions that must be met in order for a change to be considered a type-IA variation.
Such minor variations are classified in two subcategories, which impact on their submission:
The 12-month deadline to notify minor variations of type IA allows for an annual reporting for these variations, where a MAH submits several minor variations of type IA that have been implemented during the previous 12 months.
Most of these type-IA variations do not have an impact on the product information. However, in case of an upcoming submission of a variation, extension or other regulatory procedure that will affect the product information, the MAH should also include any type-IA changes affecting the product information, in order to keep the product information up-to-date and to facilitate document management.
There are no recommended submission dates for type-IA variations.. However, MAHs are encouraged to avoid submitting type-IA notifications shortly before or during the Agency holiday periods (e.g. the end of July and Christmas).
Meaning of implementation for type-IA variations
For quality changes, 'implementation' is when the company makes the change in its own quality system. This interpretation allows companies to manufacture conformance batches and generate any needed stability studies to support a type-IAIN variation before making an immediate notification 1 because the change will not be made in their own quality system until these data are available.
For product information, it is when the company internally approves the revised product information. The revised product information will then be used in the next packaging run.
1 For example, the type IAIN for addition, deletion or replacement of components in the flavouring or colouring system requires stability data on at least two pilot-scale or industrial-scale batches.
Article 7(2)(a) of the Variations Regulation sets out the possibility for a MAH to group several type-IA or -IAIN variations under a single notification to the same relevant authority, or to group them with other types of variation.
Possible grouping of type-IA and -IAIN changes only
Possible grouping of type IA and IAIN with other types of variation
It must be noted, however, that when submitting type-IA or -IAIN variations as part of a group, the legal deadlines for submission of each variation should be respected, i.e. a type IAIN should always be submitted immediately, whether or not it is grouped with other variations, and any type-IA variations should always be submitted within 12 months following their implementation.
The Agency will review the notification within 30 days following receipt, without involvement of the Rapporteur or Co-Rapporteur.
The same principle applies whether a single or a group of Type IA/ IAIN variations is being submitted.
However, if the Type IA/ IAIN Variations are grouped with other variations (Type IB, Type II, Extension), the grouped submission will follow the review procedure and timelines of the highest variation in the group and the Rapporteur will provide an assessment report for the group. Although the Rapporteur is not expected to assess the Type IA/IAIN variations in the group the Rapporteur will confirm in the assessment report whether non-acceptance of (part of) the change(s) in the group leads to non-acceptance of the Type IA/ IAIN changes in the group.
A type IA/ IAIN variation notification should contain the elements listed in Annex IV of the Variations Regulation and should be presented in accordance with the appropriate headings and numbering of the EU-CTD format. The Commission “Variations Guidelines” further specifies which elements should be included in a Type IA/ IAIN variation notification.
In order to help MAHs ensuring that their type IA/IAIN variations are complete and correct before submitting them to the Agency, it is strongly recommended to use the pre-notification checklist before submission of any type IA or type IAIN variation. Also, in order to facilitate the completion of the application form, MAHs are advised to consult the EMA/CMDh Explanatory Notes on Variation Application Form and the EMA Practical Guidance on the Application Form for Centralised type IA and IB variations.
Type IA variations are intended to provide for a simple, rapid and efficient procedure for minor changes. The MAH should be aware that the submission of redundant information or a confusing dossier presentation will not facilitate such procedures. Similarly, deficient and missing documentation can lead to rejection of the variation. However, in exceptional cases the Agency may issue a single request for supplementary information, for which a response should be provided within 4 working days in the mandatory eCTD format for electronic submissions. Failure to provide the requested information, or submission of incomplete and/or unsatisfactory responses within 4 working days may lead to an unfavourable outcome.
The following elements should be included in a Type IA/ IAIN variation notification, as specified in the Variations Guidelines:
It should be noted that the responsibility for the quality of the submitted documentation lies with the MAH and is crucial to the overall process. The MAH is responsible for ensuring that the Type IA variation complies fully with the conditions and documentation requirements as specified in the Variations guidelines.
Grouped Type IA/ IAIN variations
A ‘high-level’ procedure number should be obtained from the Agency shortly before submission. To submit your request, raise a ticket via EMA Service Desk. Please click on “Finance Services”, then the Type of ticket request to be selected is “Request for high-level procedure or ASMF number” followed by sub-option “IG Procedure Number (Type IA grouping)” and attaching a draft cover letter.
If you do not have an EMA Account, you may create one via the EMA Account Management portal.
Please note that requesting this ‘high level’ procedure number in advance is mandatory for submissions sent via the eSubmission Gateway or Web Client since this number must be included in the eSubmission Gateway XML delivery file User interface.
For procedural matters related to a type IA/ IAIN Variation for a specific product and in order to avoid rejection, please contact the EMA Service Desk, selecting the tab “Business Services”, category “Human Regulatory”. The subcategory to be selected is “Post-authorisation - Human”, followed by the sub-option “Variation IA queries”.
For more detailed queries on technical matters please contact the EMA Service Desk.
If you do not have an EMA Account, you may create one via the EMA Account Management portal. For further information or guidance about how to create an EMA Account reference the guidance "Create an EMA Account".
Submission of Type IA/ IAIN Variation Notifications
References
The Agency will review the (grouped) type-IA and -IAIN variations within 30 calendar days following receipt. The Agency will check the correctness of the application form, the presence of the required documentation and compliance with the required conditions, in accordance with the Classification Guideline.
Day 0: Receipt of type-IA or -IAIN-variation notification;
Day 1: Start of Agency check;
By day 30: Favourable or unfavourable review outcome.
By day 30, the Agency will inform the MAH by Eudralink about the outcome of the review.
Where the outcome of the procedure is favourable and the Commission decision granting the marketing authorisation requires amendment, the Agency will inform the Commission accordingly.
Where one or several type-IA or -IAIN variations are submitted as part of one notification, the Agency will clearly inform the MAH about which variations have been accepted or rejected following its review.
Type-IA and -IAIN changes can be implemented prior to submission of the notification. However, in case of unfavourable outcome, the Variations Regulation requires the MAH to immediately cease applying the rejected variations. Please refer to 'what should I do in case of an unfavourable review outcome for my type-IA or -IAIN variation?' for further details.
It is still possible for MAHs to submit type-IA notifications prior to their implementation, particularly when the proposed changes are related to other notifications or variations requiring prior approval.
In accordance with the provisions of Article 20 of the Variations Regulation, the worksharing procedure does not apply to Type IA/ IAIN variations.
However, the submission of one or several Type IA/ IAIN variations affecting more than one marketing authorisation of the same MAH, in one notification to the same relevant authority (similar to worksharing) is possible under Article 7(2) of the Regulation – see also “Can I group the submission of Type IA/ IAIN variations? Can they be grouped with other types of variations?”
This type of grouping is referred to as 'IG' by the Agency.
A ‘high-level’ procedure number is assigned for all IG procedures submitted to the Agency. This number should be systematically obtained from the Agency shortly before submission by sending your request via EMA Service Desk. Please click on “Finance Services”, then the Type of ticket request to be selected is “Request for high-level procedure or ASMF number” followed by sub-option “IG procedure number (Type IA grouping)” or “Workshare procedure number”. The letter of intent should be attached to the EMA Service Desk ticket.
If you do not have an EMA Account, you may create one via the EMA Account Management portal.
In addition, it is also possible to group a Type IA/ IAIN variation(s) with a Type IB or Type II variation, which is submitted for a worksharing procedure. In such case, the Rapporteur will be asked to confirm whether the non-acceptance of (part of) the change(s) leads to non-acceptance of Type IA/IAIN in the group. In this case, the 'high level' cross-products procedure number for the worksharing should be obtained in like manner as for IG procedures. For further information see also Worksharing: questions and answers 'What procedure number will be given to variation applications under worksharing?'
A type-IA or -IAIN variation will be rejected when:
In such cases, the MAH should immediately cease to apply the rejected changes.
In the case of a negative outcome of a type-IA application because the conditions for type-IA variations are not met and consequently a resubmission (as a type-IB or type-II variation or an extension) is needed or because documentation is deficient, it is the MAH's responsibility to judge whether the rejected type-IA variation has an impact on the quality, safety or efficacy of the medicinal product. If this is the case, the MAH has to take appropriate action.
The Agency may ask the MAH to complete a suspected quality-defect notification form and provide a risk-assessment report on the impact of the product on the market to qdefect@ema.europa.eu within seven calendar days from the date of the rejection letter. Such requests are expected to be very exceptional. The MAH has to follow the instructions under notifying quality defects or product recalls.
For information on the fee applicable for Type IA/ IAIN variations, please refer to the explanatory note on fees payable to the European Medicines Agency. Such fee covers all authorised strengths, pharmaceutical forms and presentations of a given medicinal product.
For variations introducing additional presentation(s)/pack-size(s), each additional presentation/pack-size attracts separate fees (‘x’ additional presentations = ‘x’ separate fees). Each presentation/pack-size should therefore be declared as a separate variation on the variation application form under the section ‘Variations included in this application’.
Grouped Type IA/ IAIN variations, whether consequential or not, will each attract a separate Type IA fee.
The fee will become due on the date of receipt of Type IA/ IAIN variation notification and fees will be payable within 45 calendar days of the date of the said notification. After approximately 15 days an invoice will be sent to the applicants billing address held on the Agency’s file.
The invoice will contain details of the product and type of procedure involved, the fee amount, the financial information and the customer purchase order number associated with the procedures invoiced (if provided in the eSubmission delivery file). The Agency does not accept stand-alone notifications of purchase order numbers that are not associated with a dossier.
The Agency will charge the fee for type IA variations or grouped type IA variations at the start of the procedure, irrespective of its outcome (positive, negative or partial/full withdrawal).
In accordance with the CHMP Procedural Announcements published on 15 March 2012, MAHs are reminded that fees for type IA variations become due at the start of the 30-day procedure. Fees are charged based on what has been declared in the application form regardless of the outcome (i.e. fees apply equally for accepted and rejected scopes).
The above means that, once submitted to the Agency, modifications such as addition or deletion of type IA variation scopes are not possible. The Agency cannot accept any revised application form to change the type or number of scopes applied for as part of any submission of Supplementary Information for Type IA variations.
Type IA variations which are grouped with other type of variations/extensions or which are part of worksharing procedure will continue to be charged on conclusion of the validation of the application.
Guidance on how to pay an invoice can be found on our website.
References
For information concerning submission of mock-ups and specimens in the framework of post-authorisation procedures, please refer to Checking process of mock-ups and specimens of outer / immediate labelling and package leaflets of human medicinal products in the centralised procedure, 3.4 other post-authorisation procedures.
Any changes in the number of units of medicinal product or medical device being an integral part of the medicinal product (e.g. prefilled syringes) will trigger a different EU number.
Differentiation should be made between the addition of a presentation where the two presentations will co-exist on the market on a long-term basis versus a replacement of a presentation where the new presentation will replace the previous one (it is expected that for a certain period of time, the two presentations will co-exist on the market until the stock of the previous presentation runs out).
In principle, a replacement of one presentation by another presentation does not trigger a new EU number, unless the number of units of medicinal product or medical device being an integral part of the medicinal product (e.g. prefilled syringes) is changed.
Examples of changes in presentations for replacement, not triggering a new EU number (this is not an exhaustive list):
Examples of changes in presentations for replacement, triggering a new EU number (this is not an exhaustive list):
In case of addition, as the presentations will co-exist on the market, two packs with different contents cannot be covered by the same EU number and will be considered as different presentations.
Changes in the number of any unit (not restricted to the medicinal product) or changes in the specifications of any unit (not restricted to the medicinal product) contained in the pack will trigger a new EU number.
Examples of changes that will trigger new EU numbers (this is not an exhaustive list):
If you have any questions on any upcoming submission, please contact us by raising a ticket via EMA Service Desk, selecting the tab “Business Services”, category “Human Regulatory”. The subcategory to be selected is “Post-authorisation - Human”, followed by the sub-option “Variation IA queries” or “Variation IB queries”.
If you do not have an EMA Account, you may create one via the EMA Account Management portal. For further information or guidance about how to create an EMA Account reference the guidance "Create an EMA Account".
In the specific case of a Type IAIN Variation for an additional presentation, the new EU marketing authorisation sub-number should be requested from the Agency before implementation.
![]()
The request should be sent together with a Checklist for requesting new EU sub-numbers (type IAIN and Type IB lead procedures only) and a draft Annex A (in English only) through the EMA Service Desk, selecting the tab “Business Services”, category “Human Regulatory”. The subcategory to be selected is “Post-authorisation - Human”, followed by the sub-option “New EU number request”. The request should be made at least 5 working days in advance of the intended submission of the variation. Once a number has been allocated, this number should subsequently be included in the Annex A and product information annexes submitted together with the Variation notification.
In case the Type IA/ IAIN notification affects any of the annexes, i.e. annex A, SmPC, annex II, labelling and/or package leaflet, the affected revised product information Annexes must be submitted as follows:
The 'complete set of Annexes' includes Annex A (if applicable), I, II, IIIA and IIIB i.e. all authorised presentations (if applicable), SmPC, labelling and PL texts for all strengths and pharmaceutical forms of the product concerned, as well as Annex II. The complete set of Annexes must be presented sequentially (i.e. Annex I, II, IIIA, IIIB) as one document for each official EU language. Page numbering should start with "1" (bottom, centre) on the title page of Annex I. If annex A is affected, the document should also be provided in all EU official languages as a separate set. The 'QRD Convention' published on the Agency website should be followed. When submitting the full set of Annexes in PDF format, this should be accompanied by the completed Checklist for the submission of Type IA and Type IB (without linguistic review) product information annexes and Annex A (if applicable) - human. A user guide on how to generate PDF versions of the product information and annexes is also available.
Please be reminded that in accordance with Union data protection requirements, no personal data should be included in the annotated PIs. This applies to the English version and as well as all the other languages translation versions. Please submit annotated PIs in an anonymised format (i.e. names of the reviewers removed from the track-changes). If you do not wish to do so, please ensure that the individuals whose data is included consented to its sharing with EMA and its further sharing by EMA with third parties such as other marketing authorisation applicants, marketing authorisation holders and National Competent Authorities, as relevant. EMA expressly disclaims any liability or accountability for the presence of unnecessary personal data in the annotated PI submitted by the marketing authorisation holder.
The electronic copy of all languages should be provided as part of the variation application. Highlighted changes should be indicated via 'Tools – Track Changes'. Clean versions should have all changes 'accepted'.
Icelandic and Norwegian language versions must always be included.
The Annexes provided should only reflect the changes introduced by the Variation(s) concerned. However, in exceptional cases where MAHs take the opportunity to introduce minor linguistic or typographical corrections in the texts this should be clearly mentioned in the cover letter and in the scope section of the application form.
In addition, the section “present/proposed” in the application form should clearly list the minor linguistic or typographical corrections introduced for each language. Alternatively, such listing may be provided as a separate document attached to the application form. Any changes not listed, will not be considered as part of the variation application.
In such cases and in cases where any other ongoing procedure(s) may affect the product information Annexes, the MAH is advised to contact the Agency in advance of submission or finalisation of the procedure(s) concerned.
When the Type IA/ IAIN Notification concerns several medicinal products, the relevant complete set of product information Annexes should be included in the eCTD sequence for each product concerned.
For Type IA/ IAINvariations affecting Annex A (e.g. introduction of a new presentation), translations of the revised Annex A in all EU languages should be provided as separate documents in clean PDF format and EN tracked Word, together with the variation application. Where the variation introduces (a) new EU sub-number(s), this/these should be included in the Annex A and in the product information texts as part of the variation application (see also “How to obtain new EU sub-numbers for a Type IAIN variation concerning an additional presentation (e.g. new pack-size)”?).
Similarly, in case of a deletion of a pharmaceutical form/strength/pack-size(s), the amended Annex A and product information Annexes should be provided as part of the Variation application.
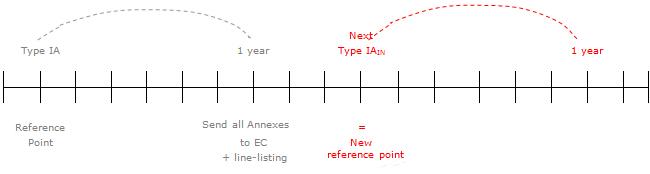
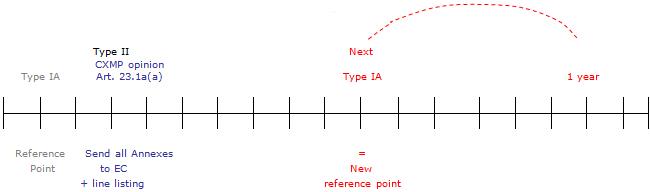
For type-IA and -IAIN variations affecting the product-information annexes to the Commission decision, the Commission decision will be updated within one year.
By the end of this period, the Agency will send the complete set of annexes, based on the latest (previously) approved annexes and reflecting the type-IA or -IAIN changes agreed during the past year together with a line-listing of those type-IA and -IAIN notifications. The Commission will subsequently issue a Commission decision on the type-IA and -IAIN notifications concerned.
However, where an opinion affecting the annexes that is followed by an immediate Commission decision, e.g. one listed in the Article 23.1a(a), is transmitted to the Commission within this yearly period, the changes of the type-IA and -IAIN notifications concerned will already be included in the annexes to that opinion and will consequently be reflected in the resulting Commission decision. This Commission decision will therefore replace the yearly updating of the marketing authorisation for the type-IA and -IAIN notifications concerned.
At the occasion of the next type-IA or -IAIN variation affecting the annexes, the procedure outlined above will be repeated based on the new reference point of the next type IA or IAIN concerned. Also see the diagram below, which illustrates the updating process.
In addition, it is important that in case of an upcoming submission of a variation, extension or other regulatory procedure that will affect the product information, the MAH should also include as a grouping application any type-IA changes affecting the product information that have not been previously notified, in order to keep the product information up-to-date and to facilitate document management.
Where a type-IA or -IAIN notification concerns several marketing authorisations, the Commission will update the marketing authorisation with one decision per marketing authorisation concerned.
Type-IA and -IAIN variations do not require prior approval before implementation ('do-and-tell' procedure), i.e. they can be implemented and notified to the Agency either immediately for type-IA variations requiring immediate notification ('IAIN') or within 12 months for type-IA variations not requiring immediate notification ('IA').
For type-IA variations affecting the product information, the date of revision of the text to be included in section 10 of the summary of product characteristics and in the corresponding section of the package leaflet at the time of printing should be the date of implementation of the change by the MAH.
The meaning of 'implementation' is explained in question 1 above ('when shall I submit my type-IA or -IAIN variation?').
If you cannot find the answer to your question in the Q&A when preparing your application, please contact us via EMA Service Desk, selecting the tab “Business Services”, category “Human Regulatory”. The subcategory to be selected is “Post-authorisation - Human”, followed by the sub-option “Variation IA queries”.
The Agency aims to respond to your query within 10 working days. To help us deal with your enquiry, please provide as much information as possible including the name of the product in your correspondence.
You should submit your query once and it is important that you submit it using the applicable type of question and sub-option. If you are uncertain on a classification of a variation as type IA or type IB please use only one of the sub-options “Variation IA queries” or “Variation IB A&B scopes queries” or “Variation IB C scopes queries”. Your query will be channelled internally to the relevant service(s) that will respond to you.
If you do not have an EMA Account, you may create one via the EMA Account Management portal. For further information or guidance about how to create an EMA Account reference the guidance "Create an EMA Account".
Type IA variations will be handled by a dedicated team of Procedure Managers (PM). You will be able to contact this PM throughout the procedure.